Analyzing Gene Lists from High-Throughput Screens
Gene lists from siRNA screens, microarray experiments, GWAS, and proteomics can be analyzed using two complementary approaches — placing genes into known biology, or discovering new biology through networks.
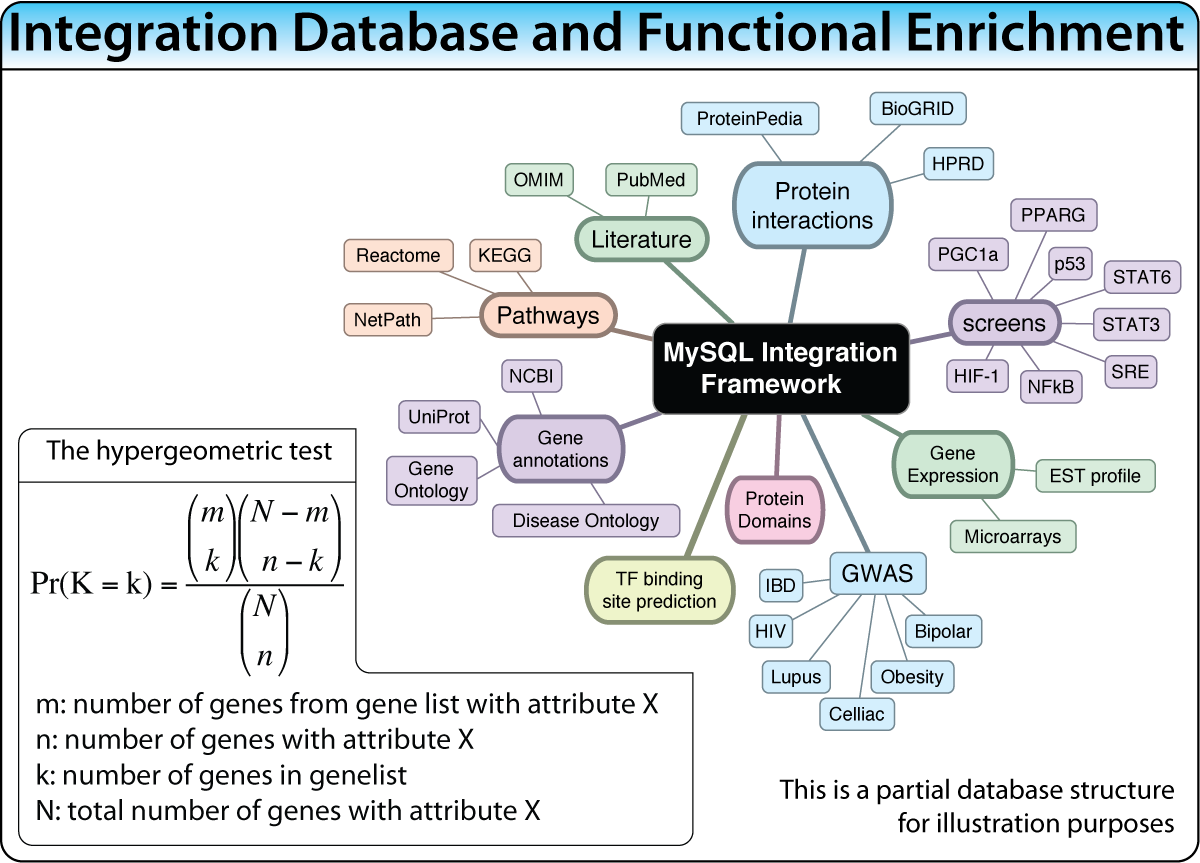
MySQL Integration Database
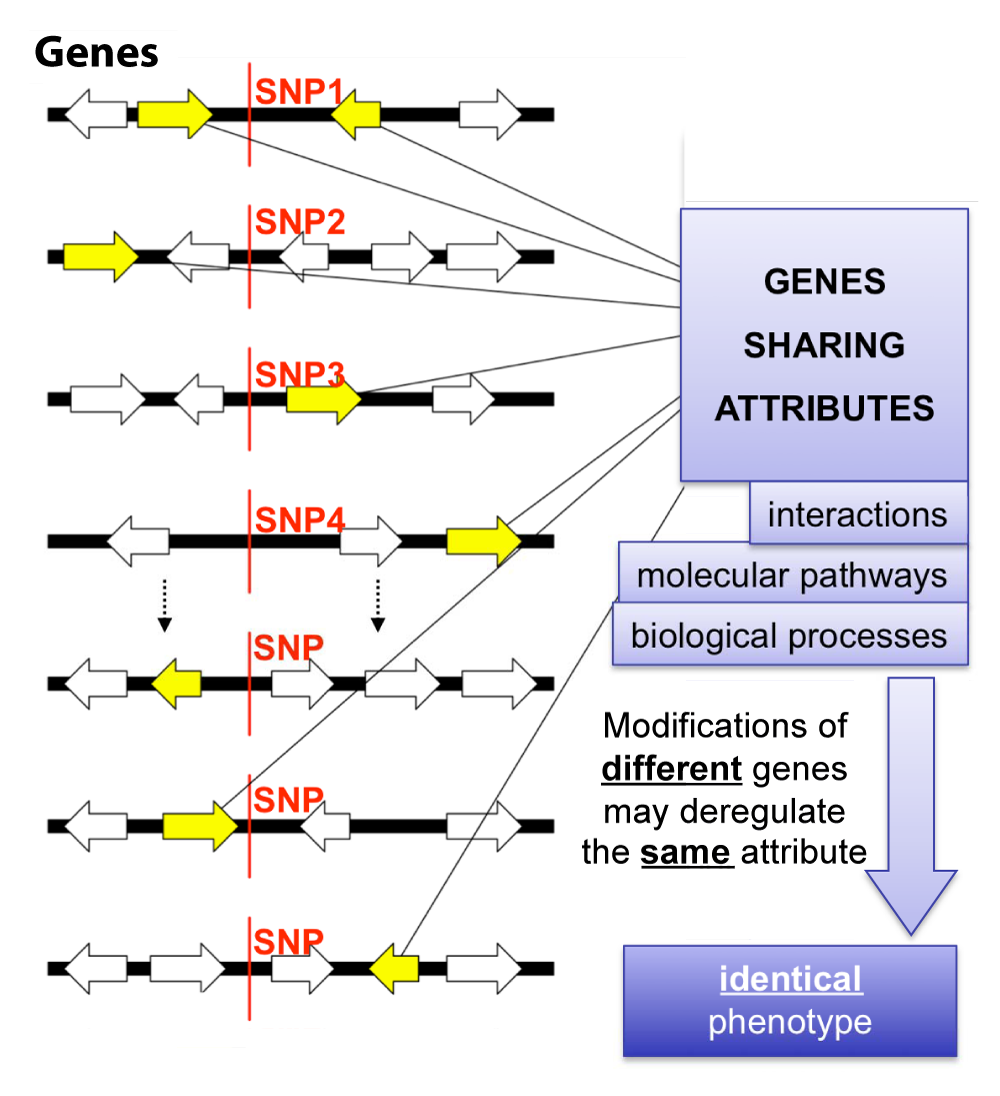
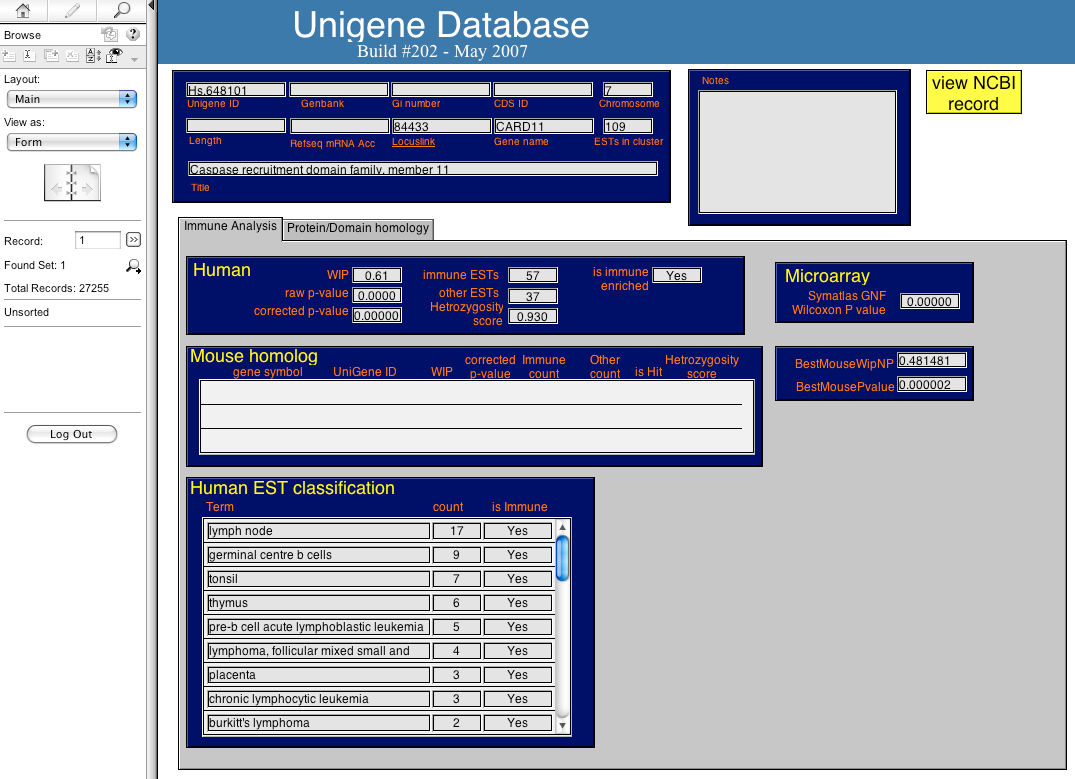
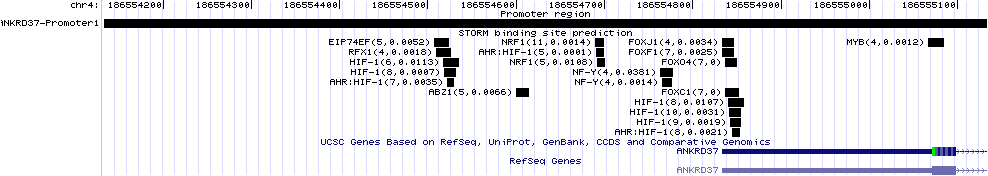
A gene-centric database framework that supports enrichment analysis and gene-gene interaction discovery. It integrates annotation from multiple sources: protein interactions, microarray expression profiles, co-citation data, transcription factor binding sites, and high-throughput screen results. Enrichment p-values are calculated using the hypergeometric distribution.
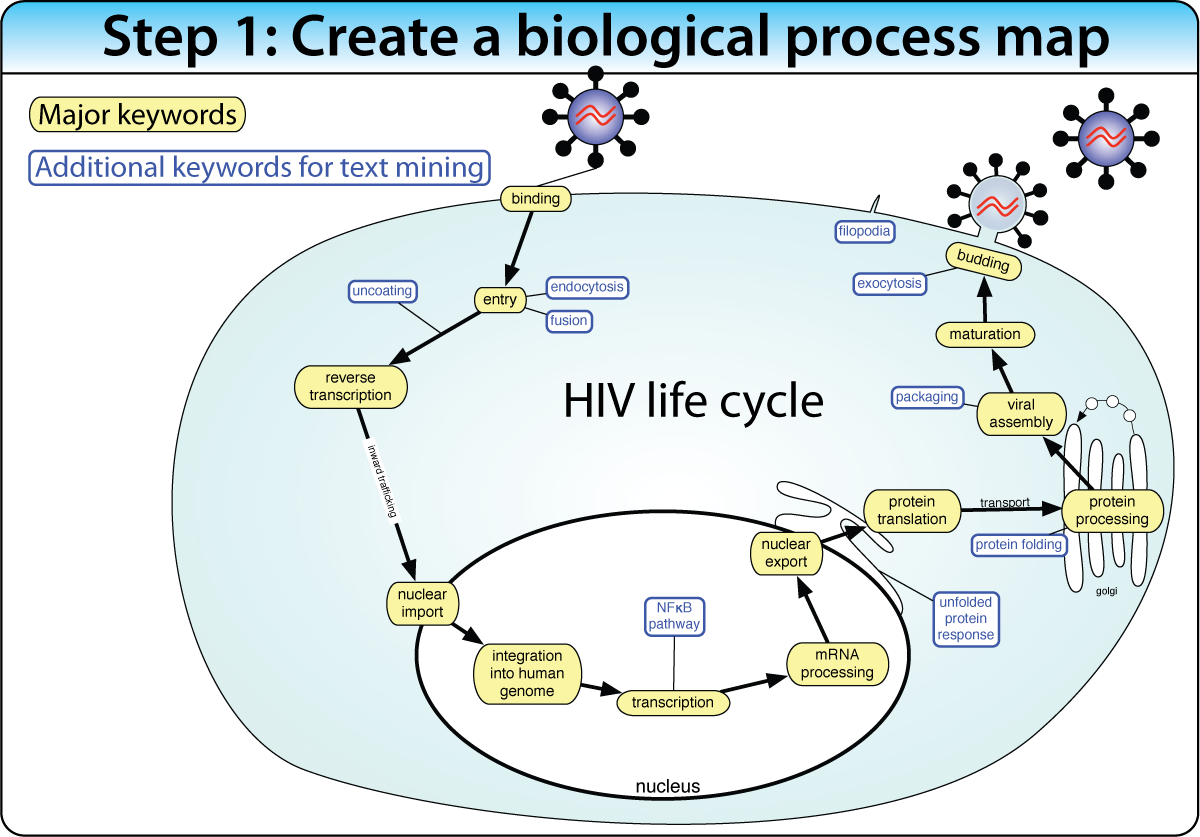
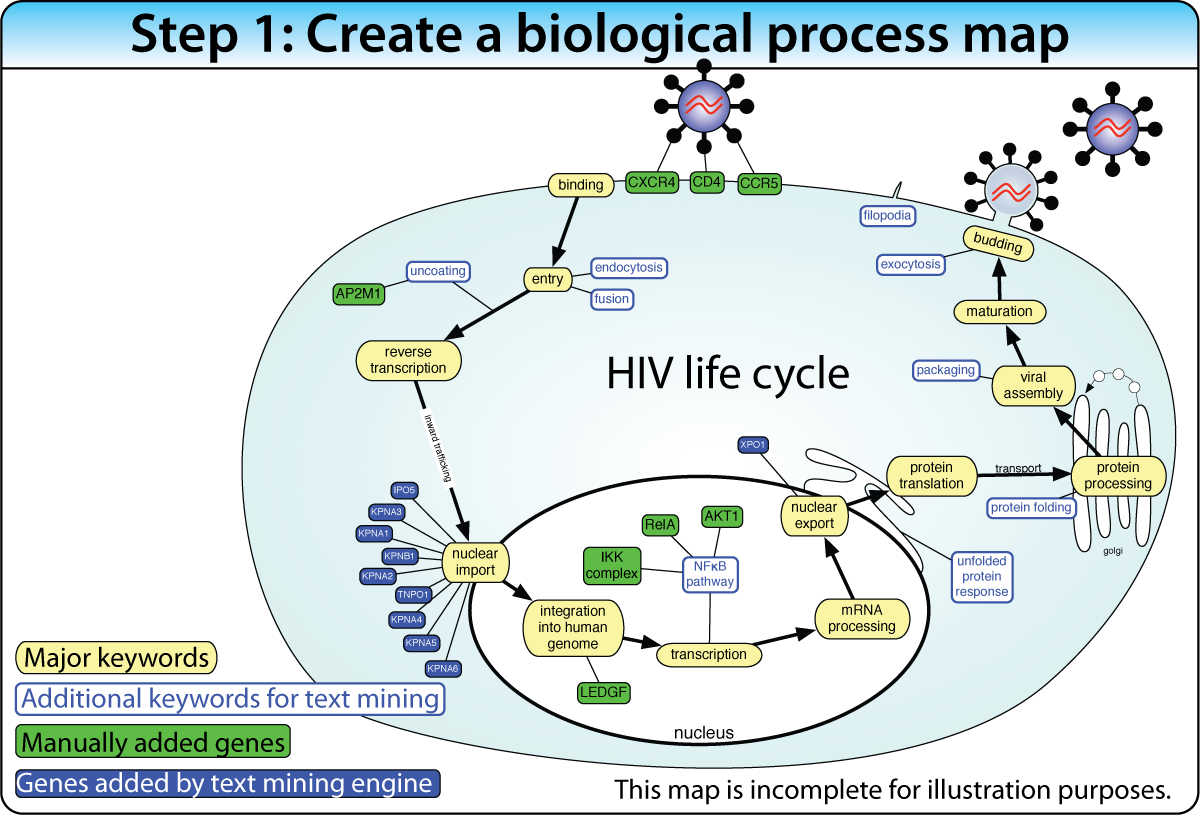
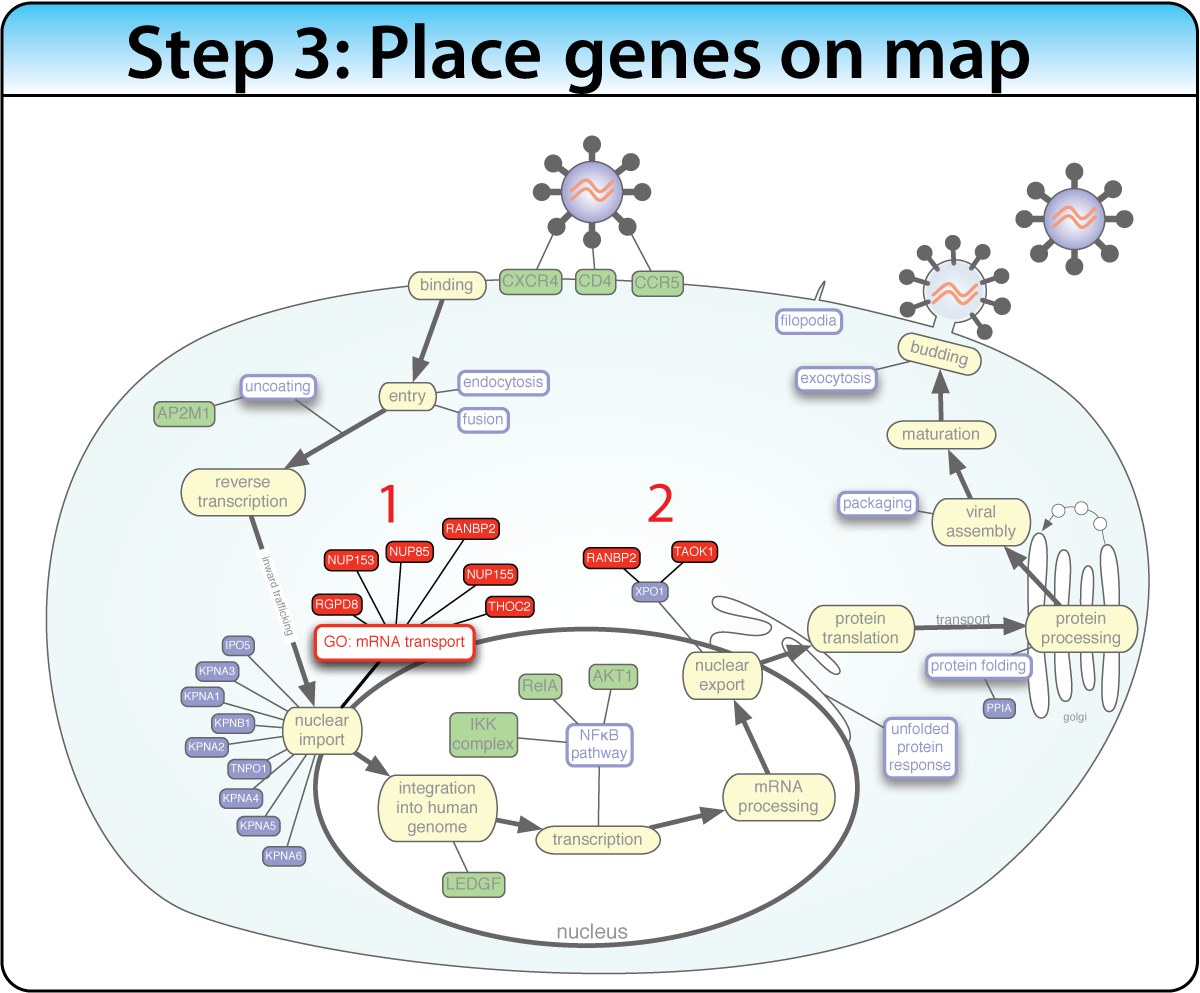
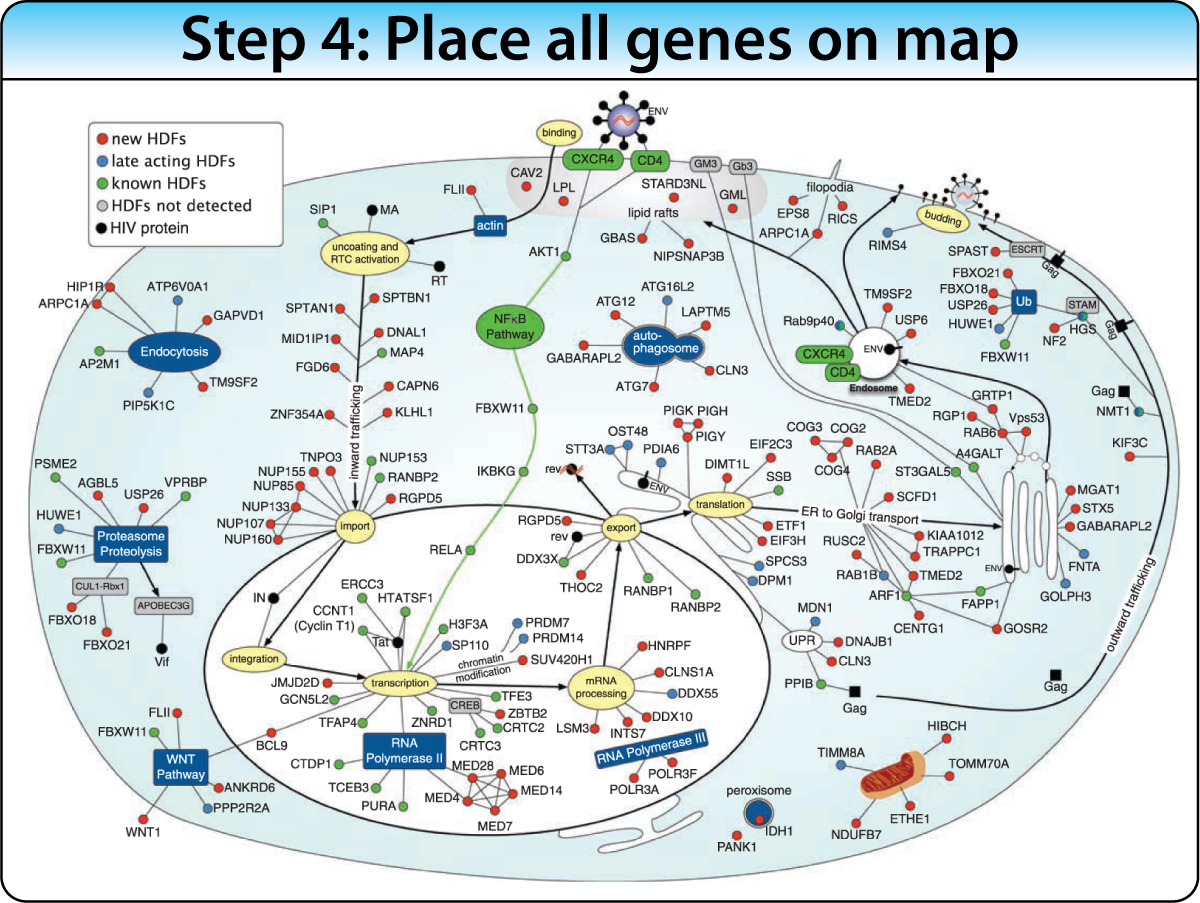
Supervised: Biological Process Mapping
Construct a biological process map of connected keywords and genes before the screen. Then place screen hits into the map using enrichment analysis and protein interactions. This approach works best when the biological pathway is well characterized.
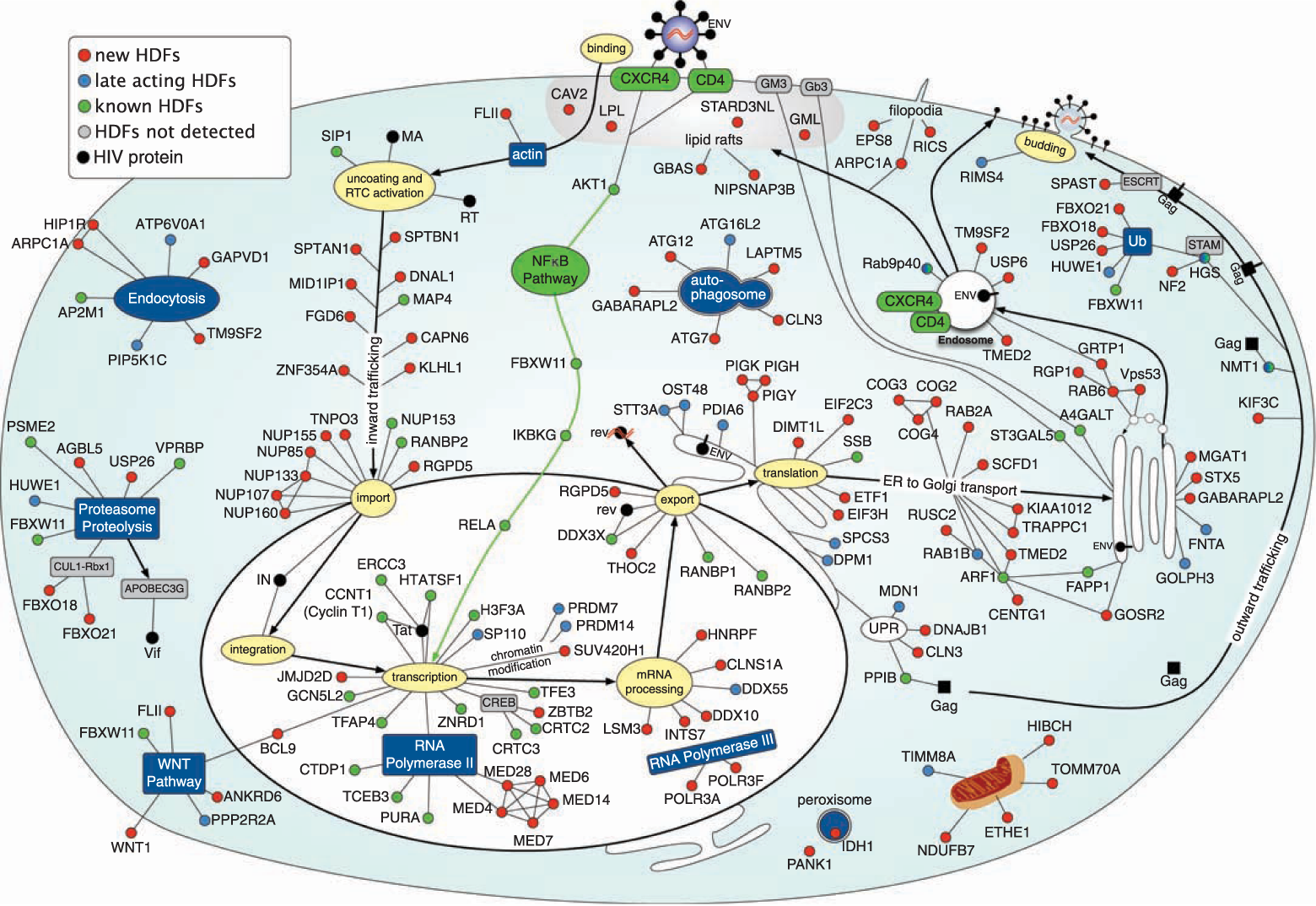
Example: Mapping 160+ genes into the HIV life cycle — from receptor binding through nuclear import to budding — using enrichment of Gene Ontology terms, protein interactions, and text mining.
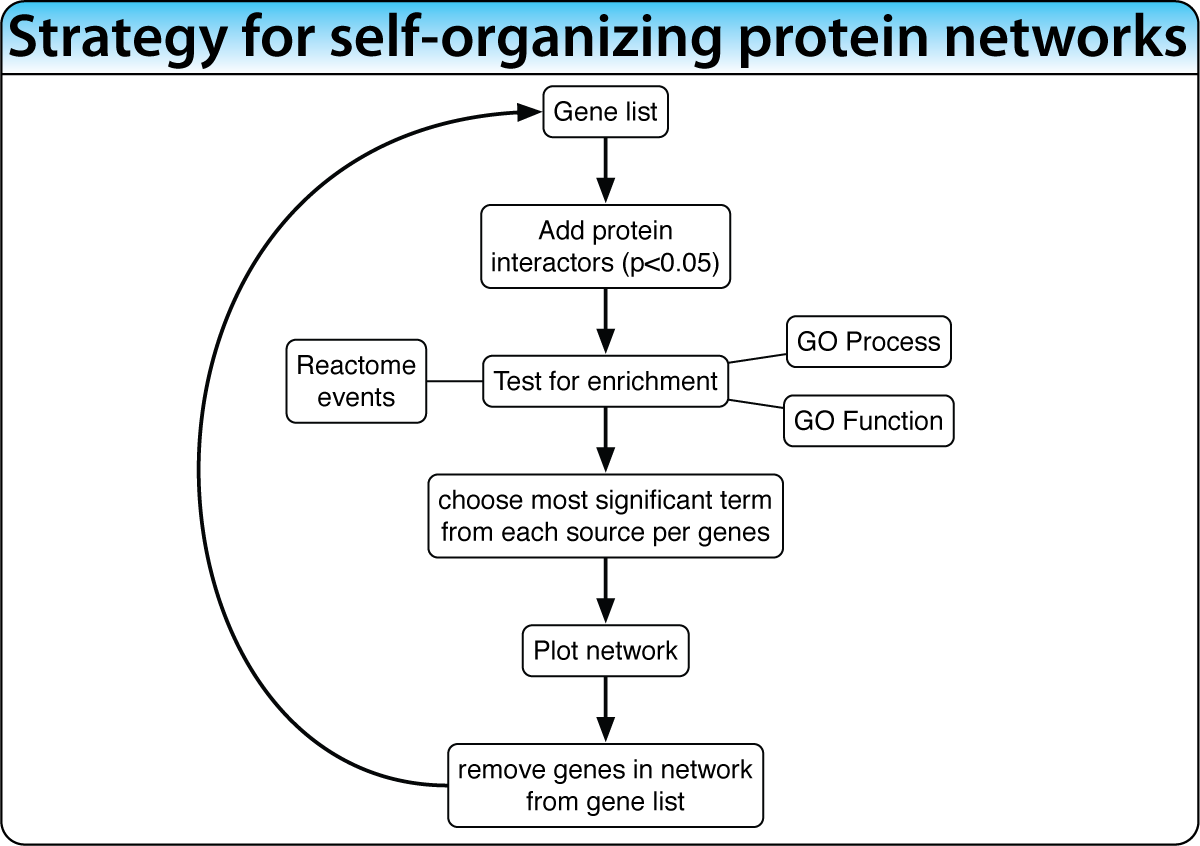
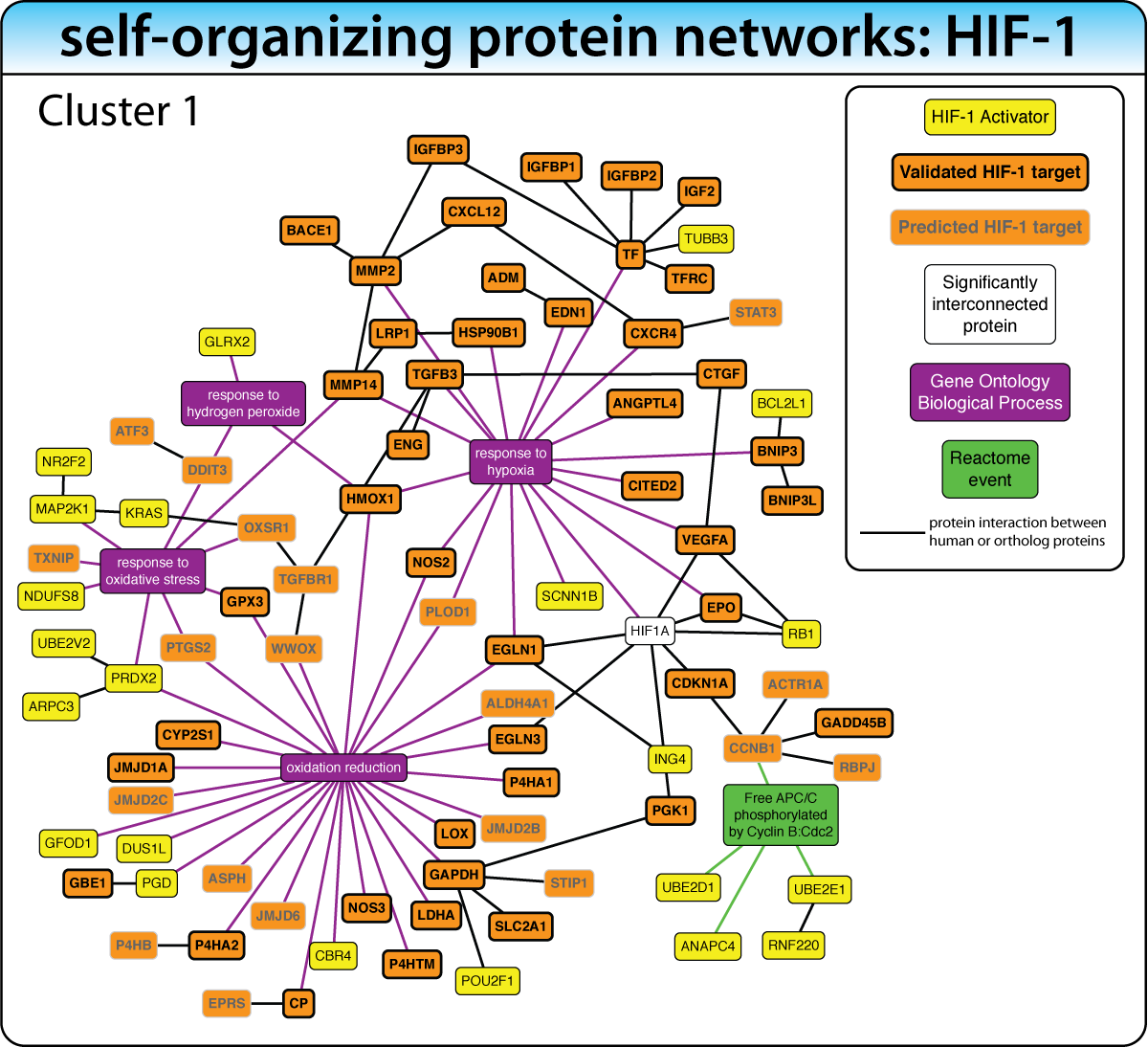
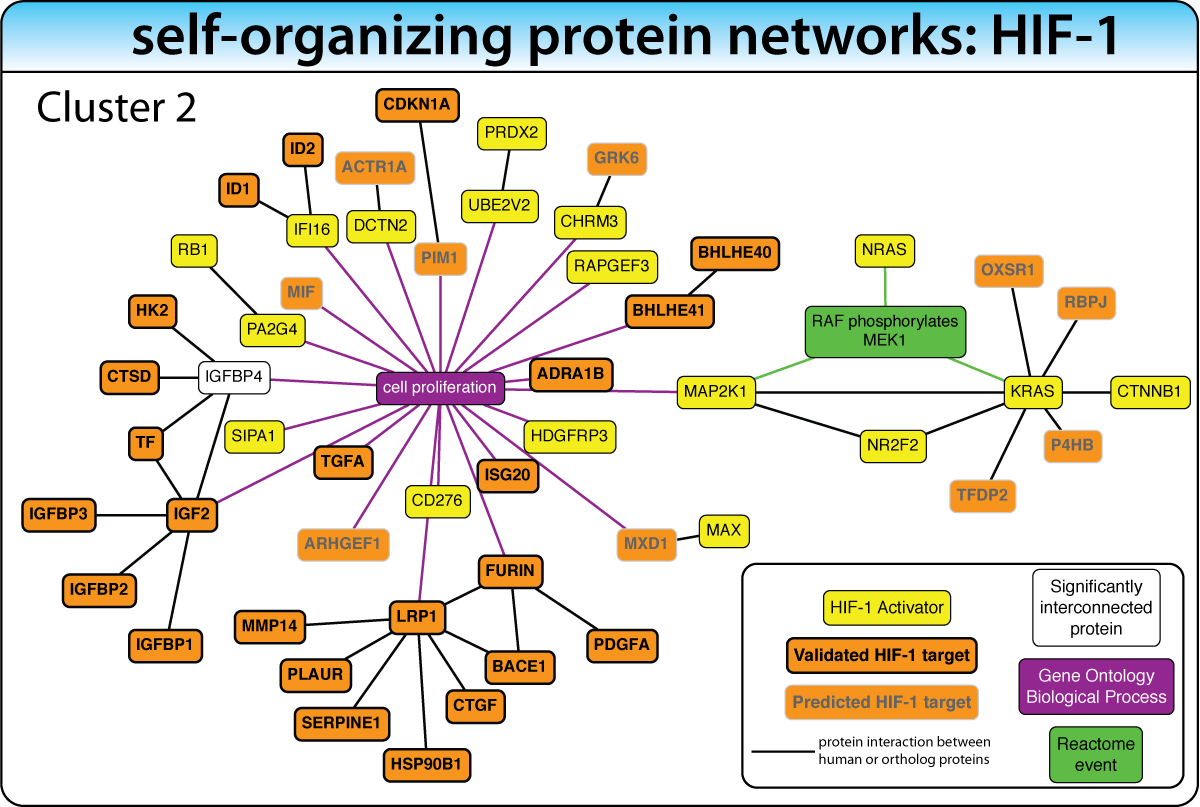
Unsupervised: Self-Organizing Networks
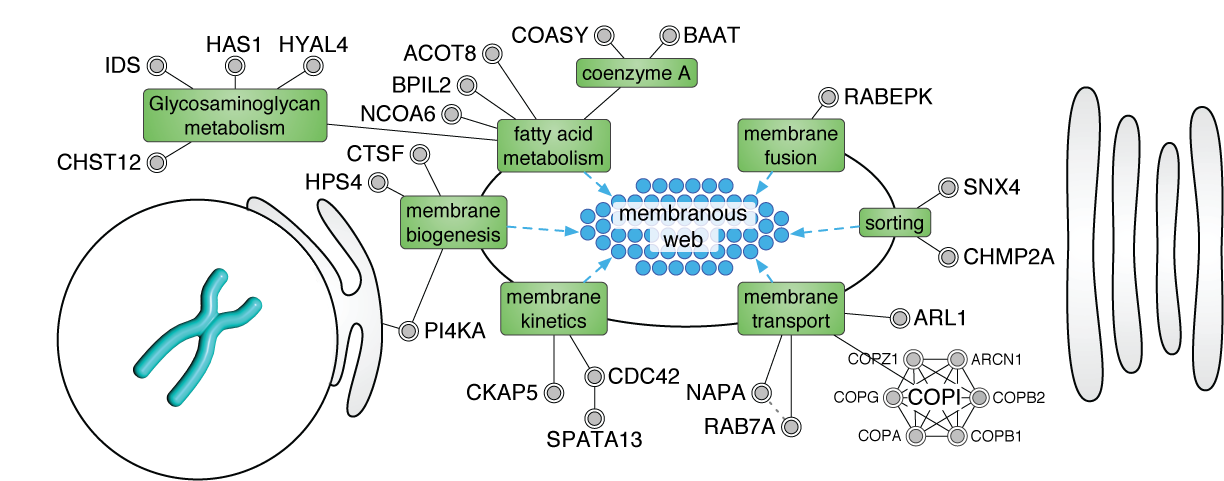
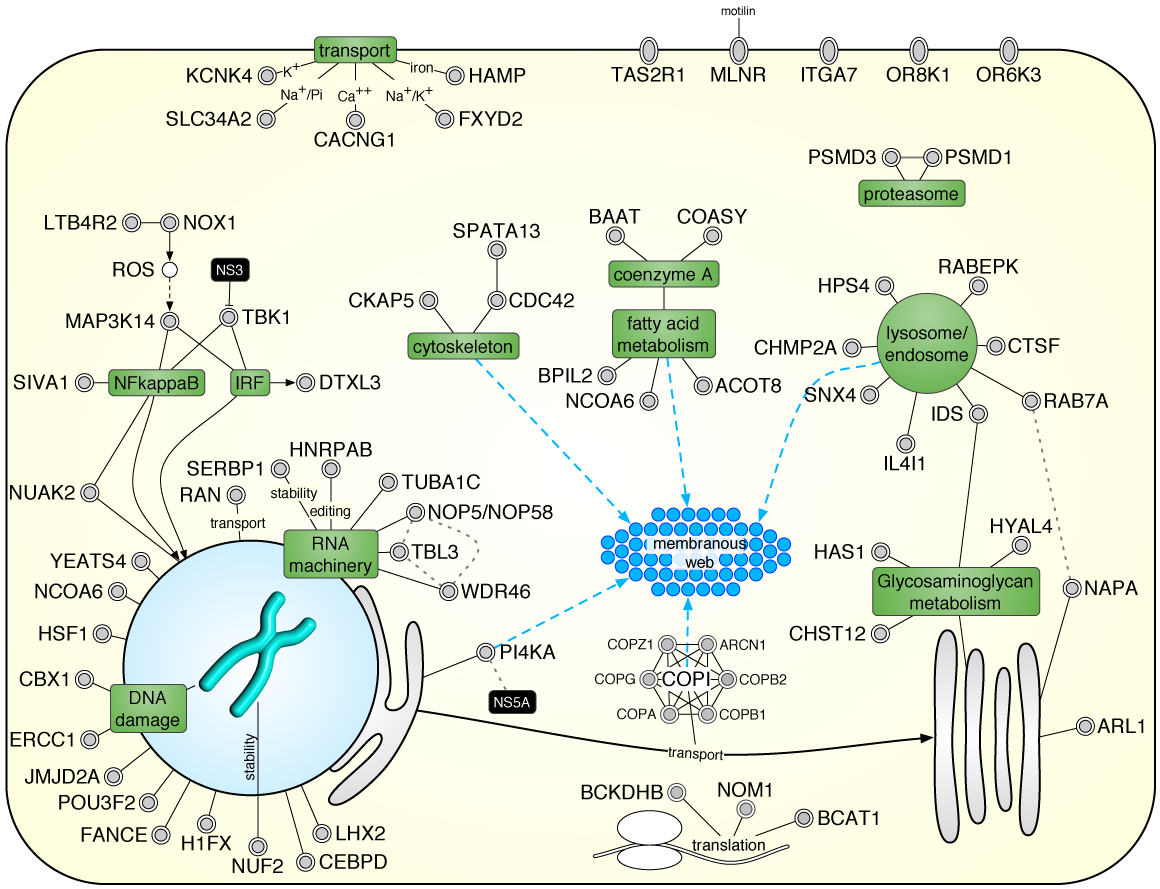
When the biological process is not well characterized, expand the gene list with enriched interacting proteins, then iteratively extract the most significant network cluster. Remove it and repeat to discover successively deeper biology.
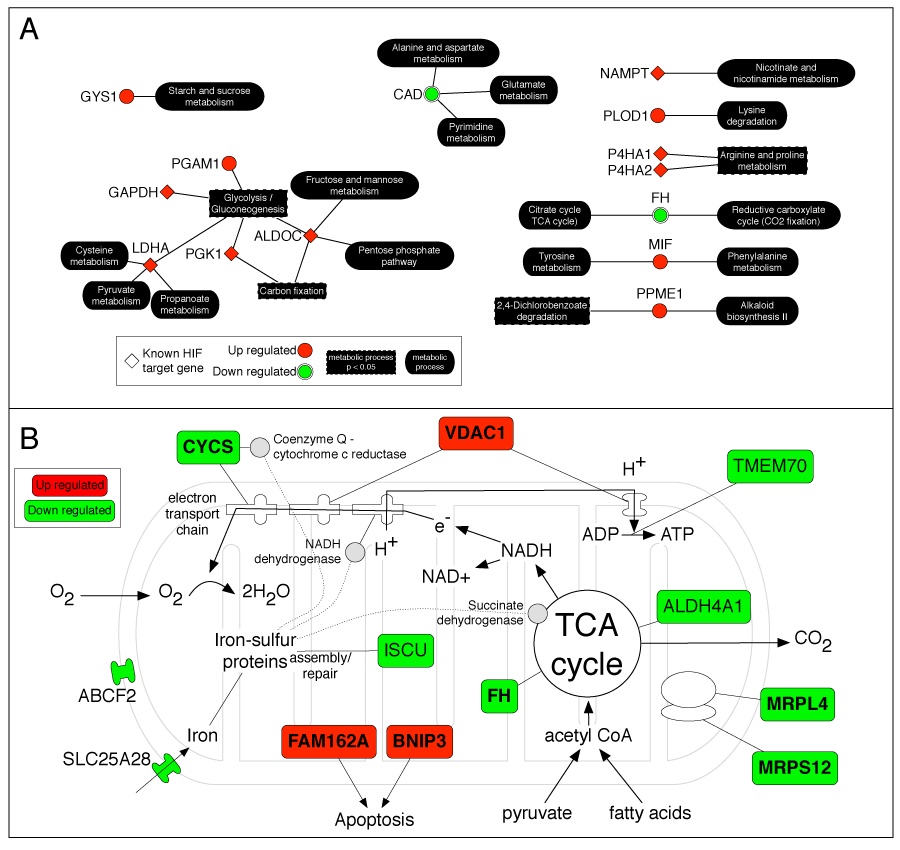
Example: In the HIF-1 screen, this strategy — biased with keywords like "hypoxia" and "oxygen" — revealed clusters for hypoxic response, cell proliferation, and metabolic reprogramming. This iterative concept is now implemented in iGEA.